Breakthrough Study Reveals Molecular Drivers of Parkinson's Disease Progression
A new scientific study reveals that the dysfunction of the parkin protein is a primary cause of toxic alpha-synuclein accumulation in Parkinson's disease. This discovery links gene regulation and protein recycling, offering new potential therapeutic pathways for managing neurodegenerative conditions.
Highlights
- •Parkinson's disease is linked to the toxic accumulation of alpha-synuclein protein.
- •Researchers discovered that parkin protein dysfunction is a key driver of this toxicity.
- •Parkin regulates essential cellular recycling pathways, including chaperone-mediated autophagy and GBA1 enzyme activity.
- •The study identifies potential therapeutic targets for restoring parkin function to treat Parkinson's and other brain diseases.
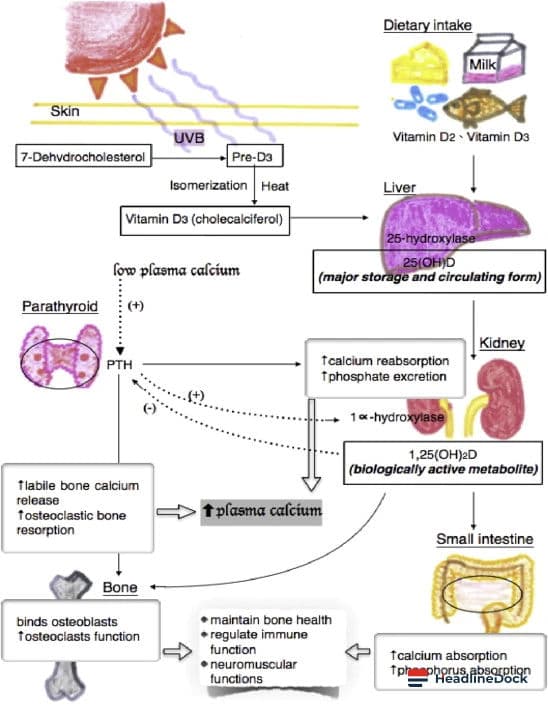
The Parkinson's disease research landscape has witnessed a significant breakthrough in understanding the molecular mechanisms underlying this neurodegenerative condition. Affecting more than 10 million individuals globally, with 200,000 cases in France, the disease is primarily characterized by the gradual loss of dopamine-producing neurons, which are vital for motor control. A new study sheds light on how a specific protein becomes toxic, offering potential new paths for medical intervention.
The Role of Alpha-Synuclein and Parkin Dysfunction
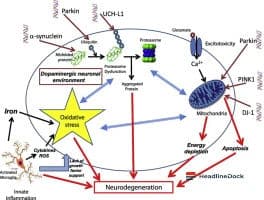
At the center of cellular dysfunction in Parkinson's disease is a protein known as alpha-synuclein. While it performs protective functions in a healthy brain, it can aggregate into toxic clumps called Lewy bodies in patients, which ultimately lead to neuronal cell death. Researchers have long sought to understand the trigger for this toxic accumulation.
Recent investigations involving collaboration with Marie-Christine Chartier Harlin's team have identified a critical mechanism linked to the failure of another essential protein: parkin. This protein serves as a versatile cellular component, acting as a tagger to mark proteins for recycling and as a regulator for specific gene activities. The study demonstrates that parkin dysfunction can directly cause the buildup of the toxic form of alpha-synuclein.
This dysfunction operates through multiple pathways. One is a direct impact on the alpha-synuclein gene. Another occurs indirectly through the regulation of glucocérébrosidase bêta 1 (GBA1), an enzyme essential for protein degradation. When parkin is inactive—whether due to genetic mutations or oxidative stress associated with aging—the expression of the GBA1 gene decreases. Furthermore, parkin works alongside GBA1 to manage chaperone-mediated autophagy (CMA), a vital protein recycling system.
Future Therapeutic Implications
When the parkin protein malfunctions, the cellular recycling system effectively breaks down. The production of protective alpha-synuclein drops, while its toxic counterpart accumulates. Researchers validated these findings by analyzing cellular models, transgenic animal studies, and post-mortem human brain samples, as well as fibroblasts from patients with parkin mutations.
These findings are crucial as they unify previously disjointed observations regarding parkin, GBA1, and autophagy into a coherent model applicable to both hereditary and sporadic forms of the disease. Looking forward, the identification of molecules capable of restoring parkin function is a primary objective. Additionally, developing specific activators to restart the deficient CMA recycling process could offer promising therapeutic strategies. These discoveries might even extend beyond Parkinson's disease, as parkin is implicated in other brain pathologies, including Alzheimer's disease and certain brain cancers.